

研究背景
锂硫(Li-S)电池是由一系列逐步转换氧化还原反应充放电的,由于其低成本、高比容量和环境可持续性,在其他电化学器件中脱颖而出,然而,锂多硫化物(LiPSs)的缓慢反应动力学、大体积波动和穿梭效应是锂硫电池商业化的主要技术障碍。与其他方法相比,如隔膜涂层,电解质调节、和负极改性、负载催化剂被认为是最直接、最有效的一种,因为它可以施加在硫正极上,调节LiPSs的限制和转化。过渡金属氧化物、过渡金属作为锂离子电池的催化剂材料被广泛研究后,往往具有较高的催化或吸附性能,这可以归因于其独特的d轨道结构。然而,考虑到Co等过渡金属的高成本问题,能否用更便宜的过渡金属取代昂贵的过渡金属,能否从废料中回收这些金属进行第二次使用?
成果简介
近日,北京大学潘锋、杨卢奕教授团队在理论预测的基础上,通过高温冲击法将废锂离子电池的废正极材料(LiCoO2和LiMn2O4)转化为Li-S电池的双功能催化剂(Co-MnO)。由于Co─MnO对多硫化物的协同催化和锚定作用,在低面积催化剂负载(<0.5 wt%)下,表现出优异的电化学性能。该工作以“Upcycling Spent Cathode Materials to Bifunctional Catalysts for High-Stability Lithium–Sulfur Batteries”为题发表在Advanced Functional Materials上。
研究亮点
(1) 通过高温脉冲加热法将废旧锂离子电池的废旧正极材料(钴酸锂和锰酸锂)转化为锂-硫电池的双功能催化剂(Co-MnO);
(2) Co催化剂有助于降低Li+扩散屏障,增强LiPSs的反应动力学,MnO催化剂与LiPSs提供强锚定位,增强分子锚定相互作用。
(3) 在低面积催化剂负载(<0.5 wt %)下,Co-MnO可以提供优异的电化学性能,包括优异的倍率性能(4 C时707 mAh g-1)和循环性能(1 C时400次循环容量衰减0.058%)。
图文导读
为了评价Co和MnO在Li-S电池中的化学吸附能力和电催化作用,进行了密度泛函理论(DFT)计算。如图1a所示,计算了聚硫化物与Co/MnO表面之间的结合能,负结合能的增强意味着化学键合能力的增强,这有利于在贫电解质条件下对LiPSs的均质化。MnO的吸附能力强于Co,表明其具有更好的锚定性能。
另外,硫转化过程中的吉布斯自由能(ΔG)值计算如图1b所示。通常,Li2S4向Li2S2的液-固转化被认为是速率决定步骤(RDS)。从剖面上看,Co表示RDS的ΔG值(0.87 eV)低于MnO(1.38 eV)。此外,MnO催化剂(0.98 eV)的步骤ΔG(Li2S2→Li2S)高于Co催化剂(0.78 eV),表明Li2S在Co活性位点上具有热力学优势。在此基础上,计算了两种衬底物上Li2S分解(Li2S→LiS-+ Li+)的能量势垒,以评价其脱氢动力学。如图1c、d所示,Li2S在Co上的分解能(0.12 eV)远低于在MnO上的分解能(2.16 eV)。综上所述,Co催化剂可以增强多硫化物的反应动力学,而Mn催化剂可以增强与多硫化物的化学吸附(图1e)。
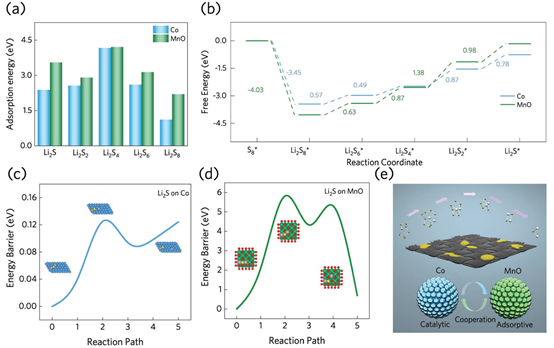
图1. (a)Co或MnO催化表面不同硫化物的结合能;(b)Co或MnO催化表面多相硫转化的吉布斯自由能谱;Li2S在(c)Co和(d)MnO表面的分解能谱;以及(e)硫转化反应的示意图。
如图2a所示,Co-MnO催化剂由废弃的LiCoO2(LCO)和LiMn2O4(LMO)正极制备。从废锂离子电池(LIBs)中收集废弃的正极,并溶解在硝酸中(两种金属的摩尔比为1:1),以获得混合的Co-Mn溶液。用碳纤维(CF)加载混合盐,然后在Ar-H2条件下进行快速热处理,截止加热温度为1500°C。快速的热激波诱导了Co和MnO粒子的形成。经过HTS处理2s后,Co和MnO颗粒均匀分布在CF上(图2b)。通过分析Co─MnO@CF的元素分布,MnO的粒径大于Co的粒径,这可能是由于Mn的熔点(1650°C)高于Co(1495°C)。利用XRD研究了各种催化剂的结构特性,如图2c所示。在26°处的宽峰可归因于CF的存在。经过HTS过程后,根据标准的PDF卡(01-077-7456和01-077-2929),可以大大识别出Co和MnO的特征峰,表明了结晶Co和MnO的形成。结合高分辨率TEM(HRTEM)图像(图2d),Co和MnO纳米颗粒分散在CF上,粒子中心的晶体结构被确定为Co(111)取向和MnO(200)取向。
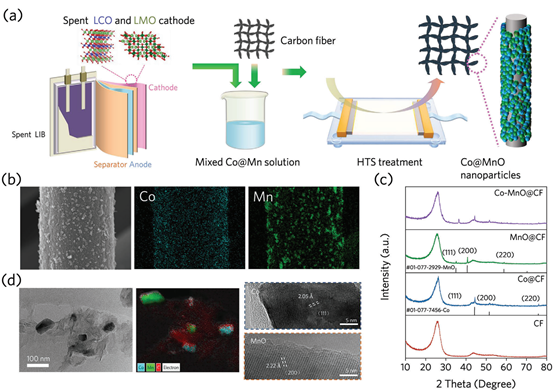
图2. (a)制备Co─MnO催化剂的工艺示意图;(b)催化元素Co和Mn的SEM图像和能量色散光谱仪映射图像,比例尺= 2 μm;(c)各种催化剂的XRD图;(d)Co-MnO催化剂的HRTEM图像。
为了研究不同催化剂的电催化性能,对含硫碳纤维(CF)、Co@CF、MnO@CF和Co-MnO@CF(分别表示为S/CF、S/Co@CF、CF和S/Co-MnO@CF)进行了一系列电化学试验。从不同正极得到的循环伏安图(CV)图(图3a)在2.28和2.01 V处出现两个还原峰,对应于S8向可溶性长链LiPSs的转变,以及进一步向Li2S2/Li2S的转变,其中,S/Co─MnO@CF正极表现出更高的峰值电流强度,表明硫的优异利用率。
此外,我们还采用了多速率CV测试来探索不同电极下的锂离子扩散速率(图3b)。根据Randles-Sevcik定律,扫描速度的平方根(v0.5)与峰值电流(Ip)呈线性相关,锂离子扩散系数(从曲线斜率(Ip/v0.5)可以得到DLi+)。S/Co─MnO@CF正极在A、B和C峰处的DLi+值分别计算为1.62×10−7、3.02×10−8和2.74×10−8 cm2 s−1,高于其他正极。此外,S/Co─MnO@CF正极在高扫描速率下表现出明显的氧化还原峰和可忽略的过电位,表明其硫转化动力学得到了改善。同时,S/Co@CF正极的锂离子扩散速率超过了S/MnO@CF阴极,说明Co催化剂更有利于促进锂离子扩散和加速反应动力学,这与计算结果一致。
为了阐明催化剂在(聚)硫化物转化的特定步骤中的催化作用,首先利用Li2S6作为活性材料组装了对称电池,如图3c所示。Co@CF的当前响应超过了MnO@CF,而Co-MnO@CF的响应最高,面积最大。这些结果表明,Co@CF比MnO具有更好的催化能力,当两种元素同时加入时,反应动力学显著增强。此外,还进一步对各种电极进行了Li2S沉淀试验,如图3d-g所示,然后根据法拉第定律分析电流-时间曲线,确定了Li2S的沉积能力。在测试材料中,Co-MnO@CF表现出最高的Li2S成核能力(148.02 mAh g−1),超过了Co@CF(125.72 mAh g−1)和MnO@CF(98.94 mAh g−1)。根据浅色条件下的峰面积直接计算容量。Co-MnO@CF具有优越的转化能力和更快的成核响应,表明Co和MnO的掺入都能有效降低初始成核过电位,加速Li2S形成的动态生成过程。此外,与MnO@CF相比,Co@CF电极表现出更大、更尖锐的成核峰,证实了Co催化剂与氧化锰催化剂相比具有更强的LiPSs还原动力学。
最后,采用线性扫描伏安法(LSV)研究了Li2S在不同电极上的氧化行为,如图3h所示。与原始的相比,Co-MnO@CF大大降低了初始电位(从−0.416到−0.581V),提高了电流响应。如图3i所示,含有Co-MnO@CF的电极提供了最低的Tafel梯度,为0.144V dec−1,表明其能够提高电子迁移能力,提高Li2S的氧化动力学。同样,Co@CF比MnO@CF具有更低的初始电位和Tafel梯度,表现出优越的反应动力学。
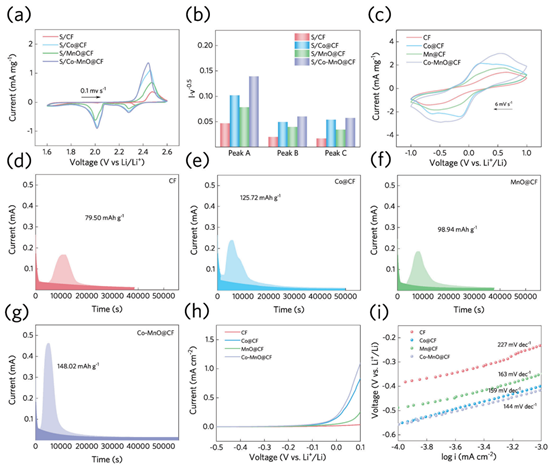
图3. (a) S/CF、S/Co@CF、S/MnO@CF和S/Co-MnO@CF正极在0.1 mV−1处的CV曲线;(b)根据S/CF、S/Co@CF、S/MnO@CF和S/Co-MnO@CF正极的多速率CV曲线计算出的峰值A、B和C处的I×v0.5值;(c)CF、Co@CF、MnO@CF和Co-MnO@CF电极的Li2S6对称电池的CV曲线;(d)CF、(e)Co@CF、(f)MnO@CF和(g)Co-MnO电极在2.05 V时Li2S成核的静电位曲线;(h)LSV图和(i)对应的四种类型电极上Li2S氧化的Tafel图。
如前所述,Co在多硫化物转化反应中表现出比MnO更优越的催化性能,而双组分催化剂则表现出最有效的整体动力学增强。因此,可以合理地推测,性能的提高源于Co和MnO之间的协同效应,其中MnO促进了LiPSs的吸附。为了进一步研究吸附行为,在恒流循环过程中对负极进行了原位拉曼分析(图4)。在开路电压下,在任何电池中均未检测到活性物质或LiPSs对应的信号。对于采用S/CF正极的电池,当释放到2.3 V的平台时,S8 2−(215 cm−1)和S6 2−(399 cm−1)在拉曼光谱中出现峰。然后,S4 2−(200和443 cm−1)和S6 2−的解离产物(─S3−,533 cm−1)出现在2.1 V的平台上。
在1.6~2.6V的充电过程中,LiPSs的峰值强度和位置保持不变,说明裸CF遭受了活性物质的不可逆损失,最终导致正极容量的显著衰减。对于具有S/Co@CF正极的电池,在放电过程中出现较弱的LiPS峰,并在充电过程中逐渐减弱,这说明Co的吸附能力可以缓解硫化物穿梭的吸附。而对于S/MnO@CF,则仅出现了较弱的S6 2−及其解离产物峰,这可能与LiPSs对MnO有较强的吸附倾向有关。最后,尽管S/Co─MnO@CF电池的吸附能力稍弱,但它表现出与S/MnO@CF相似的行为。因此,可以推测,在Co存在的情况下,尽管S/Co─MnO@CF中MnO的含量仅为S/MnO@CF的一半,但反应中的LiPSs可迅速转化为硫化锂,从而有效减少LiPSs的形成和穿梭剂量。总之,MnO和Co的联合作用可以提高正极的催化和吸附性能。
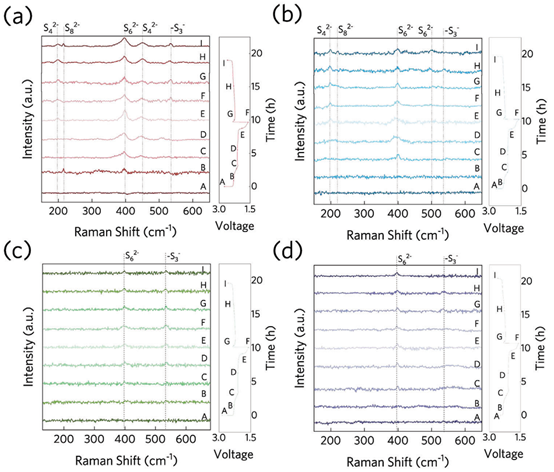
图4. 在电流密度为0.2C的情况下,S/CF、S/Co@CF、S/MnO@CF和S/Co-MnO@ CF正极的Li-S电池的原位拉曼光谱。
然后,通过恒流循环试验评价了各种催化剂的电催化性能。如图5a所示,S/Co─MnO@CF正极在1C下提供的793 mAh g−1的初始容量高于S/CF(615 mAh g−1),S/Co@CF(725 mAh g−1),和S/MnO@CF(691 mAh g−1),此外,经过400次循环后,S/Co─MnO@CF的容量保留率为76.9%,对应于每个周期的衰减率为0.058%,这也显著优于S/CF(0.18%)、S/Co@CF(0.093%)和S/MnO@CF(0.12%)。S/Co@CF正极的容量在循环过程中逐渐下降,表明在转化反应中反应物的持续损失,这可能是由于LiPSs在Co。相比之下,S/MnO@CF正极的比容量在初始循环中显著下降,并在200次循环后稳定下来。可以推测,缓慢的反应动力学可能导致了循环初期长链LiPSs的过度积累,导致了MnO上活性位点的饱和。因此,这些LiPSs将不可避免地穿梭到负极,导致容量损失。在消耗了多余的LiPSs后,剩余的LiPSs可以在MnO@CF上进行可逆转换。
为了获得更多关于活性物质在正电化学过程中的转化反应的信息,我们研究了恒流充放电过程的比容量-电压分布图(图5b)。S/Co─MnO@CF正极在充放电期间的过电位(ΔV)最低(146 mV),表现出其优越的动力学特性。电化学阻抗谱(EIS)的四个正极100循环(图5c)表明,S/Co─MnO@CF正极最小的Rct和最大的斜率,表明Co和MnO共同加速电荷的转移在电极表面,促进离子扩散过程。
在从0.5C到4C的不同倍率下,进一步评估了正极的速率性能(图5d),S/Co─MnO@CF的电池比其他电池在0.5C、1C、2C和4C具有更高的比容量(分别为904、821、764和707mAh g−1)。Co-MnO的高电催化能力和LiPS吸附能力协同加速了LiPSs的高速率转化反应,有效地锚定LiPSs并减少电池的过电位。此外,与最近的文献(如图5e所示)进行比较后发现,S/Co─MnO@CF正极的速率性能优于具有类似过渡金属或催化剂的材料。
对于商用锂离子电池,硫负荷密度也是一个关键参数。通常,实验室制备的Li-S电池具有低面积负荷和高电解质/硫比(E/S,μL mg−1),这远远低于商业上需要的能量密度。因此,我们制备了具有较高面积负荷(5.6mmcm−2)的S/Co─MnO@CF正极,E/S比控制为8。如图5f所示,在0.2C时,它的初始比面积容量为4.98 mAh cm−2,经过100个循环后,可以分别实现70.0%的容量保留,对应于每个循环的衰落率为0.3%。为了进一步证明S/Co─MnO@CF正极在实际使用中的潜力,验证软包电池与≈200μm厚的锂负极组装并进行测试(图5g)。基于S/Co─MnO@CF的柔性Li-S袋电池在弯曲180°时,初始放电容量为899 mAh g−1,在0.2C下进行100次循环后,容量保留率为67.4%。此外,我们还对与高温超导技术相关的能源消耗进行了评估,并对电极中过渡金属的可行性进行了经济分析。最终的研究结果表明,上循环策略降低了能量消耗,同时在所获得的电极中实现了更高的过渡金属利用率。这些结果表明,S/Co─MnO@CF正极在未来的便携式电子产品中具有巨大的应用潜力。
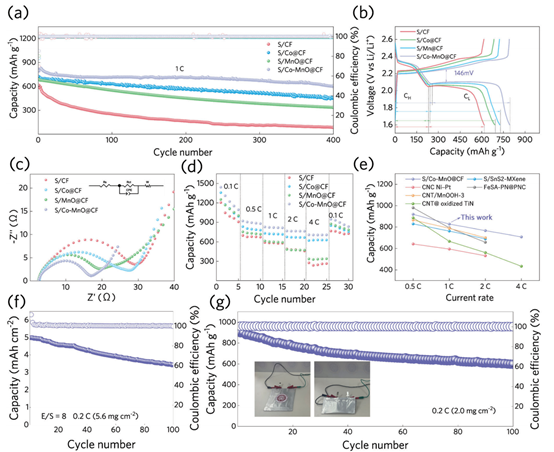
图5. S/CF、S/Co@CF、S/MnO@CF和S/Co-MnO@CF正极的电化学性能。(a) 在1C下的循环性能;(b)第一次循环的恒流充放电曲线;(c)在1C下进行100个循环后的EIS谱;(d)倍率性能的比较;(e)S/Co─MnO@CF正极的放电容量与其他类似材料的比较。(f) S/Co─MnO@CF正极在5.6 mg cm−2下的循环性能;(g)S/Co─MnO@CF软包电池在0.2C 2.0mg cm−2下弯曲180°后的循环性能。
总结与展望
综上所述,我们开发了一种高温热冲击法,将锂离子电池中的废正极转化为锂硫电池的Co-MnO催化剂。原位表征证实,Co催化剂有助于降低Li+扩散屏障,增强LiPSs的反应动力学,MnO催化剂与LiPSs提供强锚定位,增强分子锚定相互作用。结果表明,S/Co─MnO@CF正极表现出良好的循环稳定性,在1C的400次循环中,低容量衰减率为0.058%,在5.6 mg cm−2的硫负载下,实现了面积容量为3.46 mAh cm−2的100次稳定循环性能,展示了其在高能锂硫电池中的潜力。该策略为锂离子电池催化剂的设计提供了新的方向,为Ni-Co-Mn三元材料等废正极的重复利用提供了有效的途径。
审核编辑:刘清
全部0条评论

快来发表一下你的评论吧 !

